Understanding SMA Disease: A Comprehensive Overview
Spinal Muscular Atrophy (SMA), often referred to in German as Spinale Muskelatrophie, is a group of rare, inherited neuromuscular disorders that lead to the progressive loss of motor neurons, the nerve cells in the spinal cord and brainstem that control voluntary muscle movement. While some might encounter or search for the term "Smr Erkrankung" in relation to various health conditions, it's crucial to clarify that the comprehensive understanding of this particular genetic disorder centers firmly on SMA. This debilitating condition impacts a person's ability to walk, eat, and breathe, presenting significant challenges for both patients and their families. Over the past few decades, scientific breakthroughs have revolutionized our understanding and approach to SMA, transforming it from a rapidly fatal condition in its most severe forms to one with increasingly hopeful treatment outcomes.
Due to the rarity of SMA, precise incidence rates can be challenging to determine, but it is estimated to affect approximately 1 in 25,000 to 75,000 live births globally. In countries like Germany, it is estimated that between 1,000 and 1,500 individuals live with SMA, highlighting the importance of raising awareness and fostering early diagnosis and intervention. This article aims to provide a comprehensive explanation of SMA disease, delving into its symptoms, the underlying genetic causes, and the burgeoning landscape of treatment options available today.
What is Spinal Muscular Atrophy (SMA)?
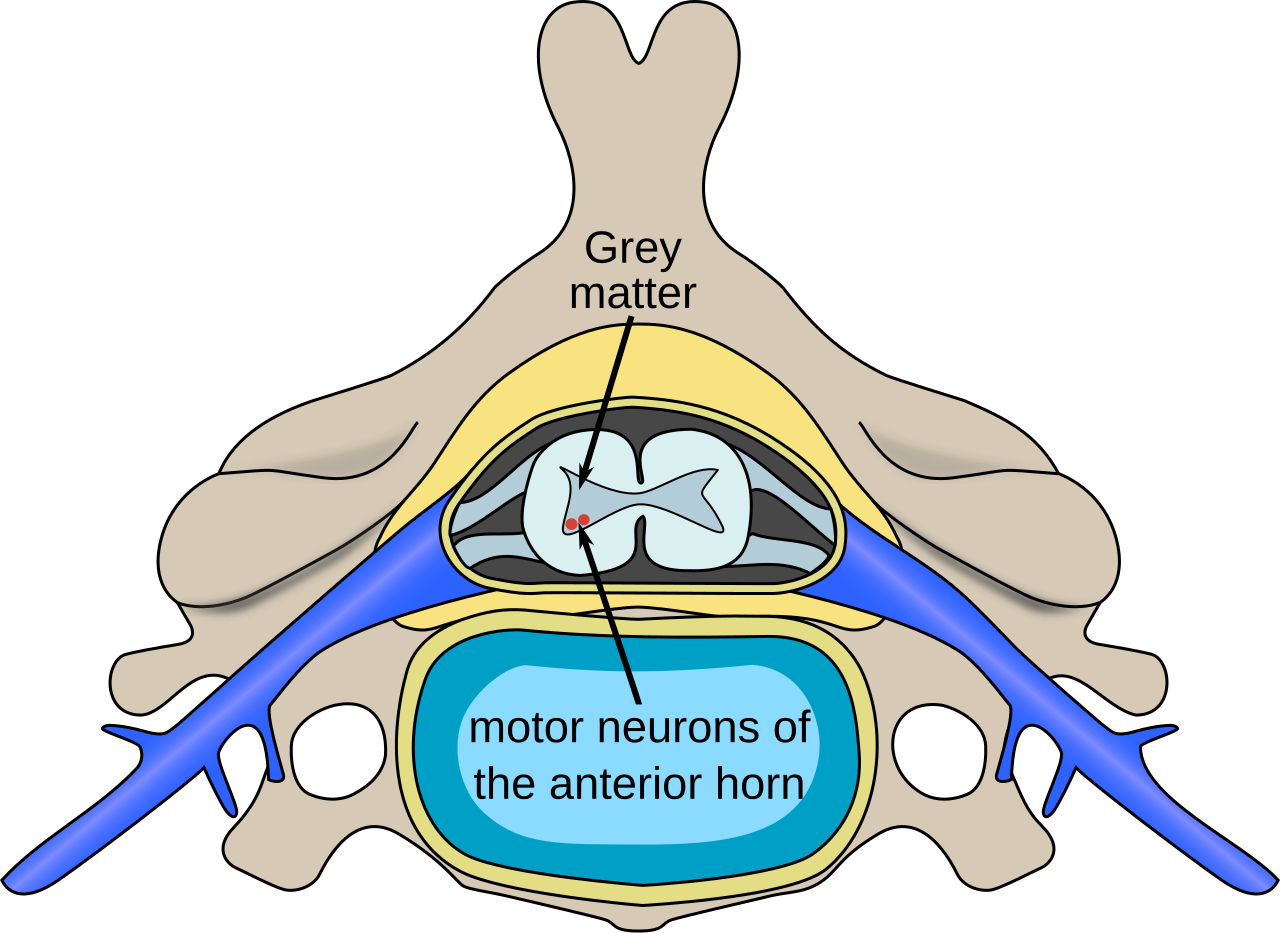
At its core, Spinal Muscular Atrophy is characterized by the degeneration of motor neurons located in the spinal cord. These critical nerve cells are responsible for transmitting signals from the brain to the muscles. When motor neurons die, the muscles they control begin to weaken and waste away, a process known as atrophy. This progressive muscle weakness affects various parts of the body, particularly the limbs, and often extends to muscles involved in crucial functions such as swallowing and breathing.
SMA is not a singular disease but rather a spectrum of conditions, with varying severities and ages of onset. This variability means that while some individuals may experience severe symptoms shortly after birth, others might not show signs until later in childhood or even adulthood. The impact of SMA can range from mild muscle weakness to severe, life-threatening paralysis, underscoring the importance of understanding the specific type and prognosis for each affected individual. Historically, the most severe forms of SMA were associated with a grim prognosis, but advancements in medical science have brought unprecedented hope, fundamentally altering the trajectory of the disease for many.
Unveiling the Symptoms of SMA: A Spectrum of Challenges
The symptoms of SMA primarily revolve around muscle weakness and its consequences, but their presentation can differ significantly based on the disease type and severity. Generally, symptoms become apparent in childhood, disrupting the normal development of motor functions. Progressive paralysis and the wasting of arm, leg, swallowing, and breathing muscles are characteristic features. For a deeper dive into the nuances of symptom presentation and the critical role of timely detection, you can refer to our detailed article: Spinale Muskelatrophie: Understanding Symptoms & Early Diagnosis.
SMA Type 1 (Infantile-Onset SMA / Werdnig-Hoffmann Disease)
- Onset: Typically within the first few months after birth, often at birth or shortly thereafter.
- Severity: The most common and severe form.
- Symptoms: Profound muscle weakness, floppy limbs (hypotonia), poor head control, difficulty swallowing and breathing, weak cry, and inability to achieve developmental milestones such as sitting independently.
- Prognosis: Without treatment, respiratory failure is common, often leading to death before the age of two due due to increasing weakness of the respiratory muscles.
SMA Type 2 (Intermediate SMA)
- Onset: Usually between 6 and 18 months of age.
- Severity: Intermediate severity.
- Symptoms: Children with Type 2 SMA can typically sit independently but usually cannot stand or walk without assistance. Muscle weakness is progressive and affects the legs more than the arms. Swallowing and breathing muscles may become involved over time.
- Associated Conditions: Due to muscle weakness, scoliosis (curvature of the spine) is common and can further impair breathing. Joint contractures (stiffening of muscles and tendons) can also develop.
- Prognosis: Patients generally reach adulthood, though they may require significant supportive care, including mobility aids and respiratory support.
SMA Type 3 (Juvenile-Onset SMA / Kugelberg-Welander Disease)
- Onset: After 18 months of age, sometimes not until adolescence or even early adulthood.
- Severity: Milder form.
- Symptoms: Individuals with Type 3 SMA can achieve the ability to walk independently, though they may experience increasing muscle weakness over time and eventually lose their ability to walk. The swallowing and breathing muscles are rarely significantly affected.
- Prognosis: Untreated, the life expectancy is typically normal, although the quality of life can be significantly impacted by progressive muscle weakness.
SMA Type 4 (Adult-Onset SMA)
- Onset: Typically in adulthood (30s and beyond).
- Severity: The mildest form.
- Symptoms: Mild to moderate muscle weakness, tremors, and twitching, primarily affecting the proximal muscles (shoulders, hips). It progresses very slowly.
- Prognosis: Normal life expectancy.
Regardless of the type, monitoring motor milestones and recognizing early signs of weakness are paramount for prompt diagnosis and intervention.
The Genetic Blueprint: Understanding the Cause of SMA
The root cause of SMA lies in a specific genetic defect. Individuals with SMA lack a functional copy of the survival motor neuron 1 (SMN1) gene. This gene is crucial for producing the SMN protein, which is essential for the health and survival of motor neurons. Without sufficient SMN protein, motor neurons degenerate, leading to the characteristic muscle weakness.
Fortunately, humans also possess a "backup" gene called SMN2. While SMN2 can produce some SMN protein, it is typically less efficient than SMN1, producing only a small amount of the fully functional protein. The number of SMN2 gene copies an individual has plays a critical role in determining the severity of SMA. Generally, the more SMN2 copies present, the more functional SMN protein is produced, which tends to correlate with a milder disease course. This genetic nuance is a cornerstone of both diagnostic and therapeutic strategies for SMA.
SMA is an autosomal recessive genetic disorder, meaning that a child must inherit two defective copies of the SMN1 gene (one from each parent) to develop the condition. Parents who each carry one defective copy are known as carriers and typically do not show symptoms themselves. Understanding this genetic inheritance pattern is vital for family planning and genetic counseling. For a deeper understanding of the genetic mechanisms, inheritance patterns, and the latest research in genetic therapies, explore our article: Genetic Roots of SMA: Causes, Types, and Emerging Treatments.
Diagnosing and Treating SMA: Hope and Progress
The journey with SMA begins with diagnosis, followed by a tailored treatment plan focused on improving quality of life and, increasingly, modifying the disease's progression. Early diagnosis is critical, particularly for the more severe forms, as early intervention can significantly alter the disease's natural course.
Diagnosis
The definitive diagnosis of SMA is made through a genetic test conducted on a blood sample. This test identifies the absence or mutation of the SMN1 gene and can also determine the number of SMN2 copies, providing crucial prognostic information. In a significant step forward, many regions have implemented newborn screening programs for SMA. This allows for the earliest possible diagnosis, often even before symptoms appear, which is vital for initiating treatment at a stage when it can be most effective in preserving motor neuron function.
Treatment Options
SMA treatment has undergone a remarkable transformation. Until recently, care was primarily supportive, aiming to manage symptoms and prevent complications. Today, several disease-modifying therapies are available, offering unprecedented hope.
Medicinal Therapies:
These groundbreaking treatments work by addressing the underlying genetic defect. They primarily focus on increasing the production of functional SMN protein, either by modifying the splicing of the SMN2 gene or by replacing the defective SMN1 gene. These therapies have shown significant success in slowing disease progression, improving motor function, and extending the life expectancy of individuals with SMA, especially when administered early.
Non-Medicinal/Supportive Therapies:
While disease-modifying therapies are crucial, comprehensive management of SMA still requires a multidisciplinary approach involving various supportive therapies. These include:
- Physical Therapy: Essential for maintaining muscle strength, flexibility, and range of motion, as well as preventing contractures and scoliosis.
- Occupational Therapy: Helps individuals adapt to their environment and maximize their independence in daily activities.
- Respiratory Support: Crucial for patients with breathing difficulties, ranging from non-invasive ventilation to more intensive respiratory care.
- Nutritional Support: Addressing swallowing difficulties and ensuring adequate caloric intake to prevent malnutrition and support overall health.
- Orthopedic Management: Including bracing, casting, and potentially surgery for severe scoliosis or other skeletal issues.
- Speech Therapy: To help with speech and swallowing challenges.
The overarching goal of all treatment approaches is to enhance the patient's quality of life, preserve existing motor function, and prevent complications, thereby enabling individuals with SMA to live fuller, more independent lives.
Conclusion
Spinal Muscular Atrophy (SMA) is a challenging neuromuscular condition, but the landscape of diagnosis and treatment has been dramatically altered by scientific advancements. From understanding its rare frequency and the spectrum of symptoms across different types, to pinpointing the critical genetic defect in the SMN1 gene and the compensatory role of SMN2 copies, our knowledge continues to expand. The advent of newborn screening and innovative disease-modifying therapies, combined with comprehensive supportive care, offers unprecedented hope for improving the lives of those affected. While the journey with SMA can be complex, continuous research, early diagnosis, and access to integrated care pathways are paving the way towards a future where the impact of this "Smr Erkrankung" (disease) is significantly mitigated, allowing individuals to thrive.